Source:
https://rwmalonemd.substack.com/p/myopericarditis-and-mrna-sars-cov
US Government political appointees and career employees knew very early, withheld the information, and blocked the minimal informed consent required by EUA

Some assert that the Cures Act is what enabled the US Federal Government to bypass informed consent regarding the gene therapy-based COVID vaccines.


Read that again.
“Except where it is not feasible, it is contrary to the best interests of such human beings, or the proposed clinical testing poses no more than minimal risk to such human beings and includes appropriate safeguards as prescribed to protect the rights, safety, and welfare of such human beings”
How does the FDA define minimal risk?
Minimal risk means that the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests.
US Government officials have repeatedly refused to provide required full informed consent for either the experimental use authorized “vaccine” products or for those participating in the clinical research and development phase of these products. One hypothesis for why this has been the case relates to the above absolution clause buried in the “21st Century “Cures” act”. If this is indeed the case, then it would seem that the FDA, CDC, DoD etc. would have a strong interest in defending the position that the "genetic vaccines” pose minimal risk to human beings. Perhaps that is why there is a pattern of chronic denialism of what are clearly significant adverse events such as myopericarditis?
Various internet researchers including Dr. Sasha Latypova and “Chief Nerd” have asserted or implied that this Cures act language is the clause which has empowered the US Government to bypass the international consensus on requirement for informed consent for medical experimentation on human subjects, all medical procedures, and administration of unlicensed medical products. But this does not appear to be the case, even though this assertion has been widely circulated on social media.
The US Government has functionally acted as the product development “sponsor” during the development and testing of the SARS-CoV-2 genetic “vaccines” under “Operation Warp Speed”. Therefore it is the responsibility of the relevant US Government employees/administrators to ensure compliance with all applicable laws and regulations involving these products during the experimental clinical research development and testing phase. Furthermore, under Emergency Use Authorization (EUA), these products are not authorized for marketing by the manufacturers, which have been acting as agents of the US Government under contract. All responsibility for the products during while under EUA vests with the relevant US Government agencies (primarily FDA, also CDC) and their administrative/Senior Executive Service and politically appointed leadership.
The FDA appears to assert, in part, that their authorization to bypass informed consent flows from a separate law, specifically Public Law 115-92 which was enacted by the115th Congress on Dec. 12, 2017, although for the life of me I cannot tell which aspects of this byzantine act confer the right to bypass true informed consent. It may be that this is another example of liberal application of administrative interpretation rather than clear congressional intent.
Here is the text relating to informed consent from the FDA guidance on Emergency Use Authorization:
b. Information for Recipients
Although informed consent as generally required under FDA regulations45 is not required for administration or use of an EUA product, section 564 does provide EUA conditions to ensure that recipients are informed about the MCM they receive under an EUA. For an unapproved product (section 564(e)(1)(A)(ii)) and for an unapproved use of an approved product (section 564(e)(2)(A)), the statute requires that FDA ensure that recipients are informed to the extent practicable given the applicable circumstances:
That FDA has authorized emergency use of the product;
Of the significant known and potential benefits and risks associated with the emergency use of the product, and of the extent to which such benefits and risks are unknown;
That they have the option to accept or refuse the EUA product and of any consequences of refusing administration of the product;46 and
Of any available alternatives to the product and of the risks and benefits of available alternatives.
Therefore, FDA recommends that a request for an EUA include a “Fact Sheet" for recipients that includes essential information about the product. In addition to the required information, the agency recommends that the content of the Fact Sheets for recipients include the following information:
Product name and explanation of the intended use of the product
A description of the disease/condition;
A description of items to discuss with a health care provider and adverse event information, including contact information for how to get more information and for reporting adverse reactions; and
Dosing information (if applicable), including specific instructions for home use or preparation (if applicable).
FDA recommends that recipients be given as much appropriate information as possible given the nature of the emergency and the conditions of the authorization.47
<Note: For the EUA recently granted for the “Booster” vaccines, there was no attempt to justify the “nature of the emergency”. Probably because there is no public health emergency, either via declaration from the Secretary of HHS or DoD (or POTUS), nor are there data indicating the current presence of a public health emergency.>
Ordinarily, FDA expects that some written form of information will be given to recipients with the MCM, similar to the Fact Sheet for health care professionals or authorized dispensers. FDA recognizes that these Fact Sheets, like those for health care professionals or authorized dispensers, will generally be brief. To ensure that individuals of varying educational levels comprehend the information provided, FDA recommends that all written information be stated in the simplest language possible using techniques to improve health literacy.48 In addition, translations to other languages may be appropriate if practicable.49 FDA recognizes that some flexibility may be needed for health care providers or authorized dispensers to make minor, nonsubstantive changes to the fact sheets for recipients such as adding local contact information, using specific letterhead or minor format changes.
Did the FDA comply with the “informed consent waiver” clauses of its own guidance on EUA? Let’s review the language:
That FDA has authorized emergency use of the product;
<Yes, this was certainly disclosed to the public>
Of the significant known and potential benefits and risks associated with the emergency use of the product, and of the extent to which such benefits and risks are unknown;
<No, actually there was not a clear and accurate statement and summary of known, potential, and unknown risks provided to citizens>
That they have the option to accept or refuse the EUA product and of any consequences of refusing administration of the product;
<No, this was not stated and in fact, the government clearly acted to directly or indirectly mandate receipt of the EUA products>
Of any available alternatives to the product and of the risks and benefits of available alternatives.
<No, the FDA actively blocked and disparaged available alternatives to the products in the form of various early drug treatment agents and products>
Based on the above analysis alone, I assert that prior and current EUA for the genetic SARS-CoV-2 vaccine products are illegal and represent arbitrary and capricious regulatory overreach.
Does the FDA have authority to issue a new EUA, or to maintain the existing EUA?
Here is the relevant guidance language:
A. EUA DECLARATION JUSTIFYING EMERGENCY USE
1. Determinations to Support an EUA Declaration
Before FDA may issue an EUA, the HHS Secretary must declare that circumstances exist justifying the authorization (section 564(b)(1)). This declaration (referred to in this guidance as an “EUA declaration”),12 must be based on one of the following actions:
A determination by the Secretary of Homeland Security that there is a domestic emergency, or a significant potential for a domestic emergency, involving a heightened risk of attack with a CBRN agent(s);13
A determination by the Secretary of Defense that there is a military emergency, or a significant potential for a military emergency, involving a heightened risk to United States military forces of attack with a CBRN agent(s);14
A determination by the Secretary of HHS that there is a public health emergency, or a significant potential for a public health emergency, that affects, or has a significant potential to affect, national security or the health and security of United States citizens living abroad, and that involves a CBRN agent or agents, or a disease or condition that may be attributable to such agent(s);15 or
The identification of a material threat, by the Secretary of Homeland Security pursuant to section 319F-2 of the Public Health Service (PHS) Act, that is sufficient to affect national security or the health and security of United States citizens living abroad.16
After the Secretary of HHS issues an EUA declaration based on one of these four determinations, and after consulting (to the extent feasible and appropriate given the applicable circumstances) with the Assistant Secretary for Preparedness and Response (ASPR), the Director of the National Institutes of Health (NIH), and the Director of CDC,17 the Commissioner may authorize the emergency use of an unapproved product or an unapproved use of an approved product, provided that other statutory criteria are met.
In appropriate circumstances, an HHS EUA declaration may support issuance of more than one EUA. For example, based on an HHS EUA declaration that circumstances exist to justify the authorization of emergency use of diagnostics for a specified biological agent, FDA may authorize emergency use for multiple diagnostic tests to meet the need, provided that each EUA meets the statutory criteria for issuance.
2. Termination of an EUA Declaration
When an EUA declaration is terminated, then any EUA(s) issued based on that declaration will no longer remain in effect.18 The HHS Secretary’s EUA declaration will terminate on the earlier of: (1) a determination by the HHS Secretary that the circumstances that precipitated the declaration have ceased (after consultation as appropriate with the Secretary of Homeland Security or the Secretary of Defense), or (2) a change in the approval status of the product such that the authorized use(s) of the product are no longer unapproved (section 564(b)(2)). For example, an EUA issued to allow an unapproved use of an approved product may no longer be needed if that product is later approved by FDA for the use permitted by the EUA.
<BOTH of these termination criteria have now been met for the mRNA-based “vaccines” from Moderna and Pfizer/BioNTech. 1) The declaration of Public Health Emergency was terminated by the POTUS (and Sec HHS) on May 11, 2023. 2) The approval status of the mRNA-based “vaccines” has changed- they are now approved. Just because the manufacturers refuse to market the approved versions in the USA (apparently due to liability issues) the FDA has in fact approved them for marketing. Therefore, based on the FDA’s own guidance, the EUA for these products is to be terminated. Issuance of a NEW EUA for the “booster” products is illegal, as there is no Public Health Emergency at this time.>
Before an EUA declaration terminates, the Secretary of HHS must provide advance notice that is sufficient to allow for the disposition of an unapproved product, and of any labeling or other information provided related to an unapproved use of an approved product (section 564(b)(3)).19
<That notice was provided, 180 days post May 11, 2023 was administratively granted to the FDA unilaterally by the FDA. Therefore, based on the sum of the above, I assert that (at a minimum) all existing EUA granted in the context of the expired COVID declaration of public health emergency shall be terminated consistent with federal law and the FDA’s own guidance when those 180 days expire- Tuesday, November 7, 2023>
What did the FDA and HHS know about the risk of Myopericarditis associated with the gene therapy-based mRNA COVID “vaccines”, and when did it know it?
In a journalistic tour de force, Zachary Stieber, Lia Onely and their colleagues at the Epoch Times have recently provided extensive documentation of who in the USG knew what and when regarding the association of myopericarditis with the mRNA-based COVID “vaccines”.
By Zachary Stieber, Lia Onely Sep 17, 2023, Updated: Sep 19, 2023
COVID-19 vaccines cause heart inflammation, U.S. authorities now acknowledge. But after being warned in early 2021 about a "large number" of cases among healthy, young people in Israel after COVID-19 vaccination, authorities did not immediately alert the public while also failing to detect a safety signal that was present in the United States, an Epoch Times investigation has found.
Even after deaths from myocarditis—inflammation of the heart—were reported and myocarditis was designated as a likely side effect of the shots, U.S. officials kept recommending vaccination for virtually the entire populace.
That led to millions of young people receiving a vaccine.
Many of those people suffered.
The CDC, America's public health agency, was warned by Israel on Feb. 28, 2021, about a "large number" of myocarditis cases after Pfizer COVID-19 vaccination, documents obtained by The Epoch Times show.
Internally, the warning was designated as "high" importance and set off a review of U.S. data. The review found 27 reported cases in the United States, according to a U.S. government memorandum dated March 9, 2021. The incidence rate was low, but "missing and incomplete data make it challenging to assess causation," the memo stated. The U.S. Food and Drug Administration (FDA), it said, "has not made a final determination regarding the causality."
Weeks later, neither the CDC nor the FDA had alerted the public to the issue, even after the death of a previously healthy 22-year-old Israeli woman and briefings from Israeli officials and U.S. Department of Defense (DOD) researchers.
Like Israel, the DOD was recording a higher-than-expected number of myocarditis cases. Patients were mostly young, healthy males.
The CDC met with military officials twice behind closed doors in April 2021. Military officials presented data during at least one of the meetings to the CDC. That presentation, which has never been released to the public, "included our preliminary patient data and analysis that suggested to us that myocarditis was indeed a possible side effect to the messenger RNA COVID-19 vaccines (within the US military)," Dr. Jay Montgomery, one of the presenters, told The Epoch Times via email.
On April 27, 2021, after the meetings, then-CDC Director Dr. Rochelle Walensky finally spoke about the matter in public, during a White House briefing.
Dr. Walensky said "we have not seen any reports" of myocarditis after vaccination. That's false, according to CDC data—the agency received 141 reports of myocarditis in the Vaccine Adverse Event Reporting System (VAERS) by the end of March 2021. Another 24 cases were recorded in the Vaccine Safety Datalink, a second system run by the CDC.
Additionally, before the briefing, Dr. Walensky was copied on multiple threads discussing myocarditis and a related condition, pericarditis, including a thread about doctors in California seeing the cases, internal emails obtained by The Epoch Times show. She responded to one of the threads, saying the information was "super helpful."
"We have not seen a [safety] signal," Dr. Walensky also told reporters during the briefing, "and we’ve actually looked intentionally for the signal in the over 200 million doses we’ve given."
Mr. Stieber has kindly also provided a detailed timeline of these and the following events which the Epoch Times’ team extensively documented and analyzed in the parent article.
Timeline: COVID-19 Vaccines and Myocarditis: A timeline of COVID-19 vaccines and myocarditis.
The results of their work are damning. The FDA and CDC were clearly aware of the scope and nature of this adverse event, and also clearly avoided disclosing what they knew to the citizens of the US and world for as long as possible. Their actions in this are the direct opposite of the informed consent information which they were required to provide by Federal law and the FDA’s own EUA guidance.
FDA and CDC administrators lied, and people (particularly children) died.
These individuals must be held accountable for their fraudulent actions. This is actual disinformation, false information intentionally distributed for political purposes. These actions clearly meet the criteria which DHS secretary Mayorkas and his colleagues have defined as domestic terrorism.
And still the FDA and CDC assert that these products are safe and effective. And still schools, athletic programs, and Universities insist that young people receive these products or they will not be allowed to participate in educational, professional certification or sports activities.
What are the practical, clinical impact of these policies? For the most recent update, I bring your attention to the comprehensive work of Mr. Ed Dowd and his colleagues and their most recent report on Death & Disability Trends, Ages 15-44: Cardiovascular Diseases, which relies on data from the UK Office of National Statistics (ONS).
In this study, we investigate the UK trends in death rates and disabilities for diseases of the cardiovascular system for individuals aged 15 to 44. We compute excess death rates and excess disability claims, which are the difference between observed deaths/disability rates and a given baseline for expected death/disability rates. We measure changes in the behaviour of morbidity and mortality before the Covid-19 pandemic with the postpandemic period for diseases of the cardiovascular system. We show a large increase in morbidity (disabilities) and mortality due to diseases of the cardiovascular from 2021. The increase in disability claims is consistent with the increase in excess deaths, and both are highly statistically significant (black swan events). The results indicate that from 2021 a novel phenomenon leading to increased cardiovascular deaths and disabilities appears to be present in individuals aged 15 to 44 in the UK.
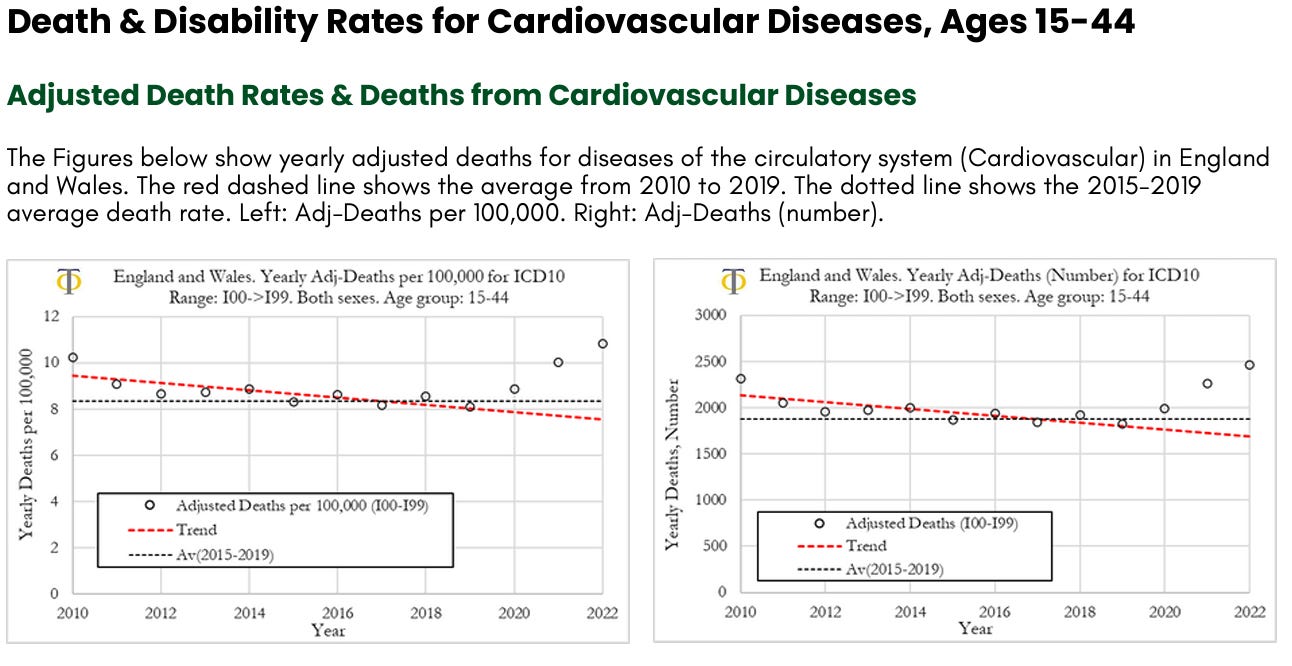
We can observe that deaths per year from cardiovascular diseases have been trending lower from 2010 to 2019, with a significant downward slope. In 2010 the deaths rate was 10 per 100,000, in 2019 it was around 8 per 100,000, a 20% drop. The death rate increased in 2020 to about 9 per 100,000 and then again in 2021 to 10 per 100,000. In 2022 the death rate increased again to about 11 per 100,000, a level that is 10% higher than observed in 2010. The death rate in 2022 was about 3 deaths per 100,000, higher than the 2015-2019 average. When translating these numbers into the absolute number of deaths for diseases of the circulatory system, shown in Figure (right), we can observe that the 5-year average deaths from 2015 to 2019 was about 1800 deaths. In 2000, cardiovascular deaths were about 2,000, 200 more than the prior 5-year average. In 2021 there were about 2300 deaths (500 more than the 2015-2019 average) and in 2022, 2500 (700 more than the 2015-2019 average).
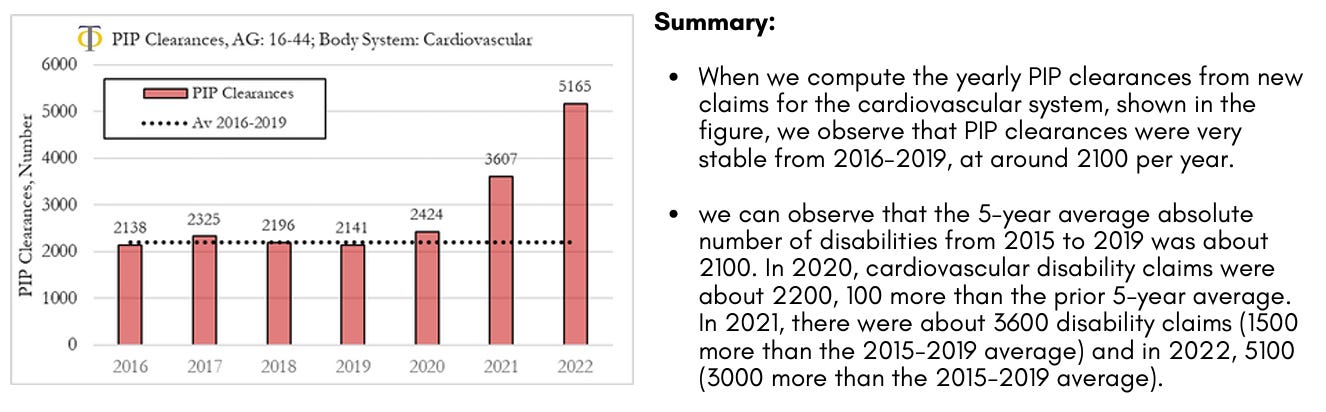
Disability PIP Clearances for new Claims from Cardiovascular Diseases
The analysis we present here refers to clearances from new claims to the system. It should be noted that clearances refer to decisions made, which can be positive or negative. The fraction of positive clearances (that lead to a grant allowance) is shown to be stable over time at a rate of about 40%. On our website, we present the analysis of trends in PIP clearances for the different body systems which include interactive charts where the user/researcher can change body system, age of the individuals and trend metric. The figure shows yearly clearances for new claims to the Personal Independence Payment (PIP) system in the UK for the cardiovascular system for ages 16 to 44. The dotted line refers to the 2016 to 2019 average yearly number of new claims.
Analysis of Excess Death & Disability Rates for Cardiovascular Diseases, Ages 15-44
Analysis of Excess Adjusted Death Rates from Cardiovascular Diseases
In this section we investigate the trends in excess deaths from diseases of the circulatory system (cardiovascular diseases), with ICD10 codes ranging from I00 to I99, in England and Wales, for the 15-44 age group. We also compare excess deaths for males and females and compute excess disability claims. In Figure below (left) we can observe that the excess deaths rates from cardiovascular diseases rose by about 13% in 2020, 30% in 2021 and about 44% in 2022. On the other hand, the excess mortality for all registered deaths UK was about 5% in 2020, 15% in 2021 and 10% in 2022. The drop in excess mortality for all registered deaths from 2021 to 2022 was not mirrored in a drop in cardiovascular deaths. The opposite occurred, with a sharp acceleration in excess deaths due to cardiovascular diseases. When looking at excess deaths for cardiovascular diseases, the Z-score in 2020 was around 3, indicating that prior to the start of the vaccinations there was already a signal pointing to an increase in cardiovascular deaths. That trend however accelerated substantially in 2021 and 2022 where we observe Z-scores of around 7.5 and 10.5, respectively. These are extreme events that we believe need a thorough investigation.
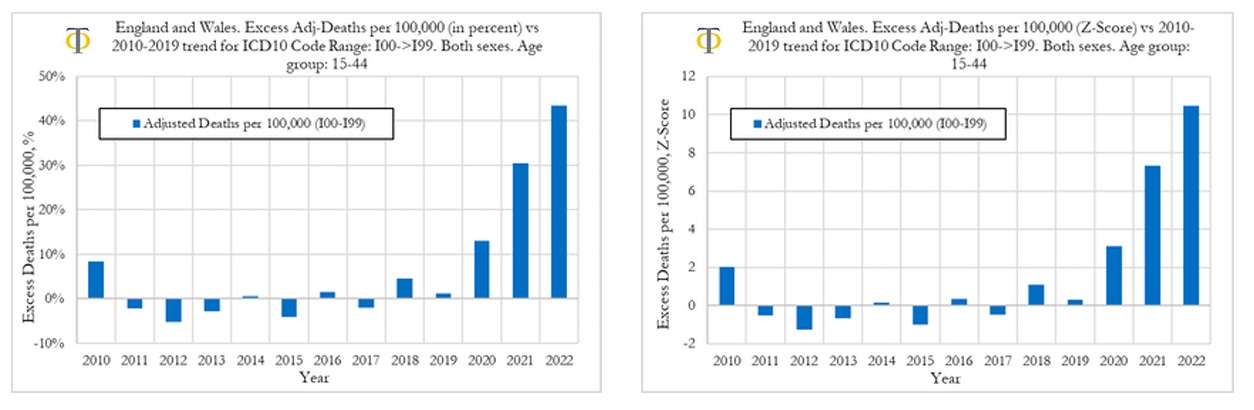
Our analysis shows that the excess death rates from cardiovascular diseases rose by about 13% in 2020, 30% in 2021, and about 44% in 2022. The excess mortality from cardiovascular deaths in 2021 and 2022 are highly statistically significant with Z-scores of 7.5 and 10.5, respectively. These are very strong signals. These signals are corroborated by similar findings when measuring rises in the fraction of deaths from cardiovascular diseases relative to all other deaths with classified causes (see full report above).
Analysis of Excess Adjusted Death Rates from Cardiovascular Diseases (Males & Females)
We now compare excess deaths rates from cardiovascular diseases for males and females aged 15-44, as shown in the Figure below. When looking at deaths attributed to the circulatory system for males and females, shown in the figure below, we observe that in 2020 and 2021 both had similar outcomes in excess mortality (deviation from trend) as well as the respective Z-scores (statistical significance). However, we also observe that in 2022 men suffered much worse outcomes than women, with men experiencing a 56% deviation from trend, compared to about 28% for women.
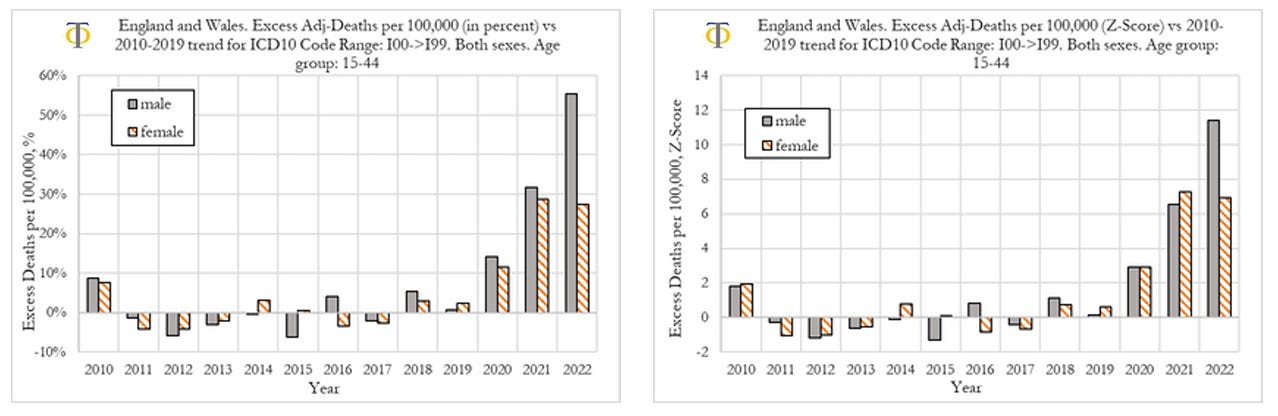
When comparing outcomes for men and women, we observe that they had similar rises in deaths from cardiovascular diseases in 2020 and 2021. However, in 2022, men suffered much worse outcomes than women, with men experiencing a 56% deviation from trend, compared to about 28% for women. Further investigation is needed. We speculate that one of the factors contributing to this difference may be men doing more physical exercise than women, which increases the probability of fatal outcomes, once the heart muscle has suffered prior (mild) injury.
Conclusions
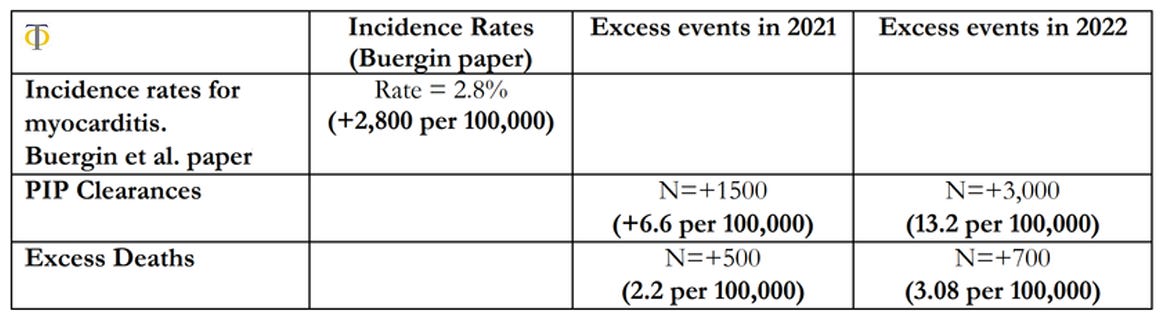
The results shown in table above indicate that there was a significant rise in both disability claims and deaths in the 15-44 age group in the UK. We observe that there was a 6.6 per 100,000 excess rate of disability claims, and only a rate of 2.2 per 100,000 excess deaths in 2021. These rates rose in 2022 with 13.2 per 100,000 disability claims and 3.08 per 100,000 excess deaths. This compares with a baseline pool of possible injured individuals of 2,800 per 100,000 reported on the paper by Buergin et al. We also observe that the relative changes in disabilities were more than double the equivalent rises in deaths, which points towards the risk of higher cardiovascular deaths in the coming years as these conditions remain unresolved. The observations above point to a worrying picture that we might see an even greater acceleration of cardiovascular deaths and disabilities in the coming years, which makes the investigation of the underlying causes of upmost importance. We are currently in the process of pursuing further investigations into this issue in more detail. In particular, we aim to analyse the trends in deaths and disabilities for the most common individual ICD10 causes within the circulatory system, in order to have insights into the underlying phenomenon of action.
And still the FDA and CDC insist that these products are “safe and effective”, and recommend them for all aged five years and over.
Who will answer for this? When will there be changes in policy, or even a modicum of common sense? By what mechanism will there be any accountability?
No comments:
Post a Comment
Note: only a member of this blog may post a comment.